What is Validation?
“…the collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality products.”
-from FDA's Guidance for Industry, Process Validation: General Principles and Practices
Call 734 274 4680 or email us at Ask VCI to find out how VCI can help you.
Validation Services Provided: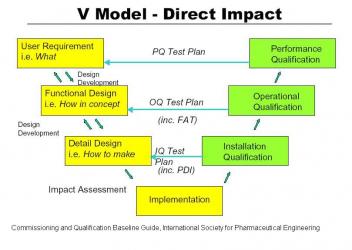
- Validation Master Plan
- Process Validation
- Process Characterization
- Computer and Automated Controls Validation
- 21 CFR Part 11, Electronic Signatures, Electronic Records
- Laboratory Validation
- Cleaning Validation
- HACCP: Hazard Analysis and Critical Control Points
Validation Master Plan.
A Validation Master Plan (VMP) is an integral part of a well organized validation project. It documents the company's approach to complex validation projects. The VMP has a broad scope. It clarifies responsibilities, general objectives, procedures to be followed for validation, and it prioritizes multiple validation tasks. It may reference several protocols and procedures to be written in order to conduct the qualification of several different pieces of equipment and different processes. It may also specify schedules for validation and the allocation of resources needed to perform the validation. Your VMP provides a means of communication to everyone associated with the project. It lets management know how the company’s resources are being allocated and when they will see the results. It tells the validation team what they have to do, when they have to do it, and gives them a means of tracking progress. Other groups can find out what the validation team is doing and what their roles are in support of the validation project. FDA can look at the VMP and realize that the validation project is well thought out and organized; that there is a logical reason for including or excluding every system from the validation project based on a risk analysis. VCI’s experience at writing Validation Master Plans can make your project go smoother whether it’s a new, greenfield plant, expansion of an existing facility, or a rearrangement of operating equipment. Click here to read “The Validation Master Plan: How to Write It and How to Make It Work for Your Company “ by Dr. Norm Howe, VCI Senior Partner, and Kristi Musgrave, VCI Senior Validation Engineer.
Process validation.
The FDA defines process validation as “…the collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality products.” Process validation is a requirement of the current Good Manufacturing Practices Regulations for Finished Pharmaceuticals (21 CFR Parts 210 and 211), Active Pharmaceutical Ingredients (ICH Q7A), and for Medical Devices (21 CFR Part 820). Validation is sometimes mistakenly viewed as a separate component that is squeezed into a gap between mechanical completion and the startup of a project. In fact it should be incorporated into the planning of the project from the outset and systems need to be in place which insure that the process will remain in compliance throughout the lifetime of the plant.
As outlined in 21 CFR 820 and in the Commissioning and Qualification Baseline Guide of the International Society of Pharmaceutical Engineers, a new project should start with a set of User Requirement Specifications (URS). These tell what the new installation is supposed to do. The URS’s should be carefully documented so that any changes can be tracked through the life of the project. A traceability matrix is a common way to accomplish the tracking of changes in User Requirement Specifications. From the URS the designers formulate a Functional Requirement Specification (FRS) which documents how the new installation is supposed to work. After review of the FRS a detail design is developed and the project is built.
Qualifications must follow an approved protocol that includes acceptable ranges and details what will happen if the tested parameters fall outside acceptable limits. There is a good reason for this. Hard experience has shown that decisions made at 3 o’clock in the morning over a recalcitrant pump can be suboptimal. When the protocol is written and approved in the calm before the storm of plant start-up, there is less of a chance that the response to an out-of-range result will be, ‘Get a bigger wrench.’
Whether your next validation project is large or small VCI can guide you through to a successful conclusion.
Process Characterization.
Before validating a process you must first characterize it. You start by defining the boundaries of the process. But the most important part is to truly understand your technology. What are the Critical Process Parameters, ie., the inputs that really determine product quality? Much of this understanding can come from your process development work and it is given to you in the design transfer documents. However, a complete understanding of the process can come only with your production equipment.
Computer and Automated Controls Validation.
Prequalification. A new project should start with a set of User Requirement Specifications (URS). These tell what the new installation is supposed to do. The URS’s should be carefully documented so that any changes can be tracked through the life of the project. A traceability matrix is a common way to accomplish the tracking of changes in User Requirement Specifications. For large automation projects like Distributed Control Systems the URS should be a separate document. For imbedded Programmable Logic Controllers it should be incorporated into the equipment URS. A loop list is generated and data backup systems are defined at this point in time. From the URS the designers formulate a Functional Requirement Specification (FRS) which documents how the new installation is supposed to work. It summarizes all activities the software will perform and should include early definitions of inputs, outputs, calculations and applications. The selection of the control system is documented. After review of the FRS a detail design is developed including loop diagrams and the IQ/OQ test plan.
21 CFR Part 11, Electronic Signatures/Electronic Records.
Part 11 provides criteria for acceptance of electronic records and signatures by FDA. It allows the use of a wide array of electronic technology. Although much confusion surrounds 21 CFR Part 11 it is in one way quite simple. Conceptually FDA wants the same security, traceability, and many other capabilities that are inherent in a paper system. For instance, if an error is discovered on a paper document we would draw a single line through the offending entry, note the correction, sign it, date it, and note down the reason for the error. FDA wants the same assurance with electronic systems. That means that the old file cannot be overwritten with the corrected data. Electronic records and electronic signatures must have the same integrity and reliability as paper records and handwritten signatures.
To what electronic data does 21 CFR Part 11 apply? Part 11 applies to all electronic records and signatures that are created, maintained, archived, retrieved, or transmitted that fall under any FDA records requirements in the Food, Drug, and Cosmetic Act, the Public Health Service Act, or the Code of Federal Regulations Title 21. These regulations are known as the Predicate Rules of which the following are of most interest for validation: Finished Pharmaceuticals (21 CFR Parts 210 and 211), Active Pharmaceutical Ingredients (ICH Q7), and for Medical Devices (21 CFR Part 820). The predicate rules mandate what records must be maintained; the content of records; whether signatures are required; how long records must be maintained, etc. If there is no FDA requirement that a particular record be created or retained, then 21 CFR Part 11 most likely does not apply to the record. VCI can help you determine which of your computer systems are subject to 21 CFR Part 11.
Part 11 regulations require controls for audit trails, system operational checks, system authority checks, metadata, and system device checks.
What is 'metadata'? It is 'data about data'. The types of metadata that can be associated with an electronic record may include: details of the record's creation, author, creation date, ownership, searchable keywords, details of the type of data found in the document, and the relationships between different data components. Metadata must be stored as an integral part of the electronic document it describes. More useful information at http://www.21cfrpart11.com/
Laboratory Validation / Analytical Method Validation.
Laboratory Validation is a process that is employed to ensure that laboratory test data and results are consistent, accurate and precise. The validation process for test methods, as well as the instrumentation that is used to perform the analysis, have IQ, OQ and PQ protocols. There are eleven main principles to the PQ laboratory test validation protocol. These points are to be applied to each and every laboratory test that is critical to the pharmaceutical manufacturing process as well as the stability program and any process validation. Not all of the eleven principles may apply to each type of testing that is performed, however, a thorough review must be done in order to ensure a complete protocol has been written. VCI's experience with a broad range of analytical methods can make your laboratory validation project run flawlessly.
The eleven PQ principles are listed below:
- Specificity
- Linearity
- Accuracy
- Precision
- Robustness
- Range
- Detection Limit (LOD)
- Quantitation Limit (LOQ)
- Ruggedness
- Selectivity
- System Suitability
Cleaning Validation.
This validation is used to show proof that the cleaning system consistently performs as expected and provides scientific data that consistently meets pre-determined specifications for the residuals.
The cleaning validation process must be written into protocols and standard operating procedures which are detailed and specific for the different pieces of equipment and instrumentation used by the facility for each type of drug product produced. Other protocols and SOP's are also required based on the type of product manufactured or process used (such as a batch or bulk process or shared versus dedicated equipment).
A final report on the cleaning validation system will attest that the studies and data prove that the process is in control and cleans as expected. This report will also detail when and why revalidation needs to take place. Call on VCI to help you clean up your cleaning validation backlog.
Hazard Analysis and Risk Based Preventive Controls (HARPC).
The HARPC process is a prevention-based food safety system. These HARPC programs are to be designed to prevent the occurrence of potential food safety problems. The system appears to be simple at first glance, however, it requires a methodical, systematic approach. A pre-requisite to a well-developed and implemented HARPC system must be a solid current Good Manufacturing Practices (cGMP) program as well as an understanding of Risk Management.
The key to the success of your HARPC program is to have your employees trained and educated on the reasons behind your HARPC plan as well as in current Good Manufacturing Practices (cGMP). Training must also be provided to your employees and management on the importance of food safety and how it applies to them. It will be essential that the unique systems of your plant and facility be considered by your HACCP team and expressed to the plant personnel.
Call VCI at 734 274 4680 to get immediate help with your Food Safety Plan or email us at Ask VCI to find out how VCI can help you.
The foundation of a sound validation plan is your Quality Management System.
The foundation of your QMS is a strong Quality Culture.